hemofilija
in Martina Feichter, medicinska urednica in biologinjaDr. med. Fabian Sinowatz je samostojni delavec v medicinski uredniški skupini
Več o strokovnjakihjaMartina Feichter je študirala biologijo pri izbirni lekarni v Innsbrucku in se tudi potopila v svet zdravilnih rastlin. Od tam do drugih medicinskih tem, ki jo še vedno navdušujejo, ni bilo daleč. Izpopolnjevala se je kot novinarka na Axel Springer Academy v Hamburgu, od leta 2007 pa dela za - najprej kot urednica, od leta 2012 pa kot samostojna pisateljica.
Več o strokovnjakihja Vse vsebine preverjajo medicinski novinarji.
Hemofilija (bolezen krvi) je motnja strjevanja krvi, ki je običajno dedna. Bolniki nimajo pomembnih faktorjev strjevanja krvi ali pa so okvarjeni. Posledično so hemofili nagnjeni k krvavitvam in zlahka modricam. Hemofilija še ni ozdravljena. Zahvaljujoč sodobnim terapijam pa lahko hemofiliki vodijo večinoma normalno življenje. Tukaj preberite vse, kar morate vedeti o hemofiliji.
Kode ICD za to bolezen: Kode ICD so mednarodno priznane kode za medicinske diagnoze. Najdemo jih na primer v zdravniških pismih ali potrdilih o nezmožnosti za delo. D66D67D68
Kratek pregled
- Kaj je hemofilija? Genetska motnja strjevanja krvi (koagulopatija). Imenuje se tudi hemofilija.
- Oblike hemofilije: Najpogostejša je hemofilija A, sledi hemofilija B. Druge oblike, kot so hemofilija C, von Willebrand-Jürgensov sindrom in parahemofilija, so manj pogoste.
- Vzrok: Pomanjkanje ali okvara faktorjev strjevanja krvi (beljakovine v krvi, ki so potrebne za strjevanje krvi). Večinoma je dedna in redko pridobljena (posledica spontane genske mutacije).
- Simptomi: povečana nagnjenost k krvavitvam, kar lahko zlahka privede do krvavitev in modric (hematomi). Krvavitev traja tudi dlje kot običajno. Kako resni so simptomi, je odvisno od resnosti hemofilije.
- Diagnostika: merjenje različnih krvnih parametrov (aPTT, hitra vrednost, plazemski trombinski čas, čas krvavitve, število krvnih ploščic), določanje aktivnosti faktorjev strjevanja krvi
- Zdravljenje: nadomestitev manjkajočega koagulacijskega faktorja (v obliki koncentratov faktorja); v nekaterih primerih druga zdravila (na primer desmopresin za blago hemofilijo A)
Hemofilija: opis
Hemofilija je prirojena motnja strjevanja krvi. Prizadeti (hemofiliki, »hemofiliki«) ne morejo tvoriti dovolj funkcionalnih faktorjev strjevanja krvi. To so beljakovine v krvi, ki so potrebne za strjevanje krvi. Zaradi pomanjkanja koagulacijskih faktorjev se krvni strdki in rane ne morejo zlahka oblikovati. Zato so bolniki s hemofilijo nagnjeni k krvavitvam. Imajo tudi rane, ki krvavijo dlje kot običajno. V določenih okoliščinah je to lahko nevarno.
Medicinski izraz za motnjo strjevanja krvi s povečano nagnjenostjo k krvavitvam je "hemoragična diateza". Splošno ime za motnjo strjevanja krvi je "koagulopatija".
Hemofilija: pogostost
Hemofilija se pojavlja skoraj izključno pri dečkih in moških. To je sorazmerno redko: le dva od 10.000 moških ima hemofilijo. V Nemčiji je prizadetih okoli 10.000 ljudi. Približno 3000 do 5000 jih ima hudo obliko bolezni.
Hemofilija: oblike
Dejavniki koagulacije so različni. Glede na to, kateri faktor strjevanja krvi vpliva na hemofilijo, zdravniki razlikujejo različne oblike bolezni.
Hemofilija A.
Pri hemofiliji A obstajajo težave s koagulacijskim faktorjem VIII (antihemofilni globulin A): telo ga ne more proizvesti v zadostnih količinah ali pa je okvarjeno. Približno 85 odstotkov vseh hemofilikov trpi za hemofilijo A. Skoraj vsi so moški.
Hemofilija B.
Pri hemofiliji B manjka koagulacijski faktor IX (antihemofilni globulin B ali božični faktor). Tudi tu so bolniki večinoma moški. V preteklosti se je hemofilija B pogosteje pojavljala v angleški kraljevi družini in v ruski carski družini. Zato ga imenujejo tudi "bolezen kraljev".
Druge oblike hemofilije
Poleg glavnih oblik hemofilije A in B obstajajo še druge dedne motnje strjevanja krvi. Eden od njih je von Willebrand-Jürgensov sindrom, imenovan tudi von Willebrandov sindrom (vWS). Tu je poudarek na von Willebrandovem faktorju: ta faktor koagulacije je premajhen ali okvarjen. Tako kot pri hemofiliji A in B to vodi v povečano nagnjenost k krvavitvam (hemoragična diateza). V Nemčiji von Willebrand-Jürgens sindrom najdemo pri do enem odstotku prebivalstva. V nasprotju z dvema glavnima oblikama hemofilije ta motnja strjevanja krvi enako vpliva na moške in ženske.
V redkih primerih se pojavijo tudi druge motnje strjevanja krvi, ki temeljijo na pomanjkanju faktorjev strjevanja krvi. Prav tako imajo povečano nagnjenost k krvavitvam (hemoragična diateza). Tej vključujejo:
- Hemofilija C: pomanjkanje funkcionalnega faktorja XI
- Parahemofilija: pomanjkanje delujočega faktorja V
- Hipoprokonvertinemija: pomanjkanje funkcionalnega faktorja VII
- Stuart Prower Pomanjkanje faktorja: Pomanjkanje delovnega faktorja X
- Hagemanov sindrom: pomanjkanje funkcionalnega faktorja XII
- Pomanjkanje fibrinaze: pomanjkanje funkcionalnega faktorja XIII
Hemofilija: simptomi
Simptomi hemofilije A (pomanjkanje faktorja VIII) in hemofilije B (pomanjkanje faktorja IX) so enaki - tudi če v vsakem primeru vplivajo na različne faktorje strjevanja krvi. Na splošno lahko rečemo: manj ko deluje faktorjev strjevanja krvi, bolj je izrazita klinična slika.
Hemofilija: stopnje resnosti
Resnost hemofilije je odvisna od tega, koliko se zmanjša aktivnost faktorjev strjevanja krvi v primerjavi z njihovo funkcijo pri zdravih ljudeh. Če se dejavnost faktorja le nekoliko zmanjša, prizadeti običajno nimajo nobenih simptomov. Po drugi strani pa je pri simptomatskih hemofilih aktivnost faktorja manj zmanjšana, kar povzroča izrazitejše znake. Obstajajo tri stopnje resnosti hemofilije (A in B):
Blaga hemofilija:Faktorska aktivnost je 6 do 45 odstotkov normalne aktivnosti zdrave osebe; približno 20 odstotkov vseh ljudi s hemofilijo ima to resnost. Prizadeti pogosto ne opazijo veliko svoje bolezni. Zato se hemofilija pri mnogih ljudeh odkrije šele v adolescenci ali odrasli dobi, ko operacije ali večje poškodbe krvavijo dlje, kot je bilo pričakovano.
Zmerna hemofilija: Dejavnost faktorja je 1 do 5 odstotkov normalne aktivnosti; prizadene tudi približno 20 odstotkov vseh hemofilov. Simptomi običajno postanejo očitni v prvih nekaj letih življenja z nenavadno dolgotrajno krvavitvijo in pogostimi modricami. Tako kot pri blagi hemofiliji je krvavitev običajno posledica poškodb ali operacije. Spontana krvavitev je redka.
Huda hemofilija: Dejavnost faktorja je manj kot 1 odstotek normalne aktivnosti. To velja za približno 55 odstotkov vseh hemofilikov. Hemofilija je običajno opazna že ob rojstvu: odstranitev popkovine sproži veliko krvavitev. Težke krvavitve iz nosu niso redke v otroštvu. Poleg tega lahko tudi najmanjše poškodbe ali izbokline povzročijo obilno krvavitev pod kožo (modrice). Pogostejša je tudi notranja krvavitev, na primer boleča krvavitev v velike sklepe (kot so kolena, komolci). Običajno številne krvavitve nimajo očitnega vzroka (spontana krvavitev).
Hemofilija: tveganja in nevarnosti
Krvavitve v sklepih (hemartroza) se pogosto pojavljajo, zlasti pri hudi hemofiliji. Posledično se lahko prizadeti sklep deformira, se prezgodaj obrabi (osteoartritis) in postopoma utrdi. Ljudje z napredovalo hemarthozo komaj premikajo roke in noge in so odvisni od invalidskega vozička.
Druga nevarnost pri hemofiliji je krvavitev v mišice: lahko poškoduje mišično tkivo in povzroči mišično oslabelost.
Načeloma se lahko notranja krvavitev pri hemofiliji pojavi na katerem koli področju organov. Krvavitve v možganih so redke, a nevarne: na primer lahko poslabšajo sposobnost razmišljanja in zmanjšajo koncentracijo. Huda možganska krvavitev je lahko celo usodna! Krvavitev v trebuhu je v določenih okoliščinah lahko tudi smrtno nevarna. Močne krvavitve v orofarinksu lahko vplivajo na dihalne poti.
Poleg nagnjenosti k krvavitvam (hemoragična diateza) lahko hemofilija katere koli stopnje resnosti povzroči tudi motnje celjenja ran.
Von Willebrand-Juergenski sindrom: simptomi
Ljudje s von Willebrand-Jürgenskim sindromom imajo tudi povečano nagnjenost k krvavitvam. Večinoma se pojavijo rahle krvavitve pod kožo (modrice), krvavitev dlesni ali dolgotrajna krvavitev po operaciji, na primer po ekstrakciji zoba. Obstajajo pa tudi bolniki, ki trpijo zaradi močne krvavitve.
Hemofilija: zdravljenje
Terapija s hemofilijo je odvisna od vrste in resnosti hemofilije. Zdravnik bo za vsakega bolnika izdelal ustrezen načrt terapije. Poleg zdravljenja z zdravili lahko priporoči tudi splošne ukrepe. Na primer, lahko je priporočljivo, zlasti v primeru hude hemofilije, telesno skrbeti in se izogibati nekaterim (škodljivim) športom.
Hemofilija A in B
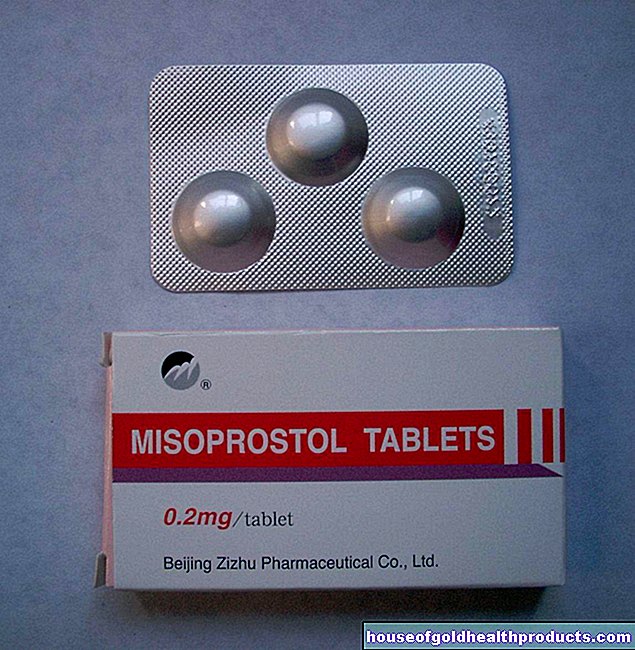
Danes so za zdravljenje hemofilije na voljo tako imenovani faktorski koncentrati. To so koncentrati faktorjev strjevanja krvi VIII (za hemofilijo A) ali IX (za hemofilijo B), pridobljeni iz krvne plazme ali gensko spremenjeni. Vbrizgati jih je treba v veno (intravensko). Mnogi bolniki se sami naučijo vbrizgati koncentrat faktorja. To jim daje veliko neodvisnosti pri spopadanju z boleznijo.
V šestdesetih in sedemdesetih letih prejšnjega stoletja je veliko ljudi v Nemčiji okuženih s hepatitisom in / ali virusom HIV zaradi kontaminiranih faktorskih pripravkov. Danes se to praktično ne more več zgoditi: krvna plazma je pred dajanjem strogo nadzorovana in predhodno obdelana. Z gensko spremenjenimi koncentrati faktorjev na splošno ni nevarnosti okužbe.
V primeru blage in zmerne hemofilije je dajanje koncentrata faktorja potrebno le, če je potrebno (zdravljenje na zahtevo): Antikoagulant se daje na primer v primeru močnejše krvavitve ali pred načrtovano operacijo. Manjših poškodb, kot so odrgnine, ni treba zdraviti s koncentratom faktorja. Krvavitev lahko običajno ustavimo z rahlim pritiskom na območje krvavitve.
Ljudje s hudo hemofilijo pa si morajo redno injicirati koncentrat faktorja (dolgotrajno zdravljenje). Faktor VIII pri hemofiliji A se daje dva do trikrat na teden. Faktor IX pri hemofiliji B je na splošno treba injicirati le enkrat ali dvakrat na teden zaradi daljšega zadrževanja v krvi.
Operacije in akutne poškodbe
Pred operacijo (tudi če je načrtovana odstranitev zoba) je treba za vse bolnike s hemofilijo opraviti pripravljalno terapijo. Običajno se v ta namen daje koncentrat faktorja. To je edini način za preprečitev resnih zapletov zaradi prekomerne izgube krvi med operacijo.
V primeru blage hemofilije A lahko namesto koncentratov faktorja pred načrtovano operacijo damo zdravilo, ki stabilizira strjevanje krvi. To na primer vključuje desmopresin. To je umetno proizvedena beljakovina. Spodbuja sproščanje shranjenega faktorja VIII iz krvnih žil. Zdravilo se lahko uporablja le nekaj dni, sicer bodo trgovine kmalu prazne.
Nasvet: Pred načrtovanim posegom bi se morali ljudje s hemofilijo s svojim hematologom ali centrom za hemofilijo pogovoriti o tem, katera preventivna terapija je v njihovem primeru priporočljiva.
V primeru akutnih poškodb v nujnih primerih je poleg lokalnih ukrepov za hemostazo (npr. Povojev za pritisk) potrebna tudi uporaba faktorskega koncentrata.
Koncentrat faktorja: zapleti
Nekateri ljudje tvorijo protitelesa (zaviralce) proti faktorjem koagulacije v koncentratu faktorja. Ta tako imenovana inhibitorna hemofilija je pri ljudeh s hemofilijo A bistveno pogostejša kot pri ljudeh s hemofilijo B. Zaviralci inaktivirajo dodani koagulacijski faktor. Terapija potem ni tako učinkovita, kot bi želeli. Hemofilija B ogroža tudi hude alergijske reakcije in druge zaplete.
Količina zaviralcev v krvi je podana v tako imenovani enoti Bethesda (BE). Višja kot je vrednost BE, več zaviralcev je v bolnikovi krvi.
Pri hemofiliji A lahko rahlo kopičenje zaviralcev pogosto nadomestimo s povečanjem odmerka koncentrata faktorja. Če se zaviralci tvorijo v velikih količinah, se priporoča imunsko tolerančna terapija: bolnik v kompleksnem režimu zdravljenja prejme zelo visoke odmerke manjkajočega koagulacijskega faktorja. Imunski sistem bi se moral postopoma navaditi na njegovo prisotnost in ustaviti nastajanje zaviralcev.
Redki inhibitor hemofilije pri hemofiliji B se obravnava drugače. Na primer, prizadeti prejemajo zdravila, ki vplivajo na imunski sistem (imunomodulatorje).
Zdravilo proti bolečinam
Huda hemofilija lahko prizadene prizadete. Na primer, krvavitev v sklepih je lahko zelo boleča. Nato pomagajo lajšalci bolečin, kot je ibuprofen. Nasprotno pa protibolečinska acetilsalicilna kislina (ASA) ni primerna za hemofilijo: povečuje nagnjenost k krvavitvam (stranski učinek ASA).
von Willebrand-Juergenski sindrom (VWS)
Pri von Willebrand-Jürgenskem sindromu razlikujemo med različnimi vrstami, ki se zdravijo različno: Pri tako imenovanem tipu 1 se aktivna sestavina desmopresin daje po potrebi (pred operacijo ali v primeru akutne krvavitve). Spodbuja sproščanje shranjenih koagulacijskih faktorjev.
Desmopresin se uporablja tudi pri tipu 2; vendar zdravilo tukaj ne deluje vedno. Nato zdravnik namesto tega predpiše koncentrat faktorja (z von Willebrandovim faktorjem).
Bolnike z VWS tipa 3 vedno zdravimo s koncentratom faktorja.
Hemofilija: vzroki in dejavniki tveganja
Hemofilija je prirojena genetska motnja, ki je običajno dedna. Redkeje se pojavi spontano (kot posledica spontane spremembe gena = spontana mutacija).
Pri hemofiliki so genetski podatki, potrebni za tvorbo funkcionalnega koagulacijskega faktorja, napačni: pri hemofiliji A je to faktor VIII koagulacije, pri hemofiliji B faktor IX. Posledica napačnega načrta gradnje je, da ustreznega koagulacijskega faktorja ni mogoče proizvesti v dovolj funkcionalni količini. To moti strjevanje krvi: rane se ne zaprejo tako hitro, zato krvavitev traja nenavadno dolgo. Nekateri bolniki imajo tudi spontano krvavitev (brez očitnega vzroka).
Hemofilija A in B: dedovanje
Načrti (geni) za vse dele telesa so na kromosomih. V jedru vsake celice v telesu je 46 kromosomov, med njimi dva spolna kromosoma, ki med drugim določata spol. Ženske imajo dva X spolna kromosoma (XX): Vsak X kromosom je podedoval od matere in enega od očeta. Moški pa imajo Y -kromosom, podedovan od očeta, in X -kromosom, podedovan od matere (XY).
Geni za koagulacijske faktorje so na kromosomu X. Pri ženskah, ki imajo dva kromosoma X, če eden od njih vsebuje napačen načrt koagulacijskega faktorja, se to običajno lahko nadomesti z drugim kromosomom X. Zato so vse življenje brez simptomov.
Če ima takšna ženska otroka, enega izmed dveh X kromosomov posreduje potomcem. Verjetnost, da gre za kopijo z napačnim načrtom, je 50 odstotkov. S prenosom genetske napake mati postane nosilec (nosilec) hemofilije. Če obstaja sin, se bo rodil kot hemofilik. Po drugi strani pa hči običajno postane potencialna nosilka.
V redkih primerih ustrezni koagulacijski faktor ni dovolj oblikovan niti pri ženskih vodnikih. Rane zaradi poškodb ali operacij lahko nato krvavijo dlje časa. Še bolj redko je, da dekle podeduje okvarjen kromosom X od obeh staršev. To je večinoma povezano s Turnerjevim sindromom dedne bolezni. Prizadeta dekleta kažejo celotno sliko hemofilije.
Von Willebrand-Juergenski sindrom
Pri von Willebrand-Jürgenskem sindromu navodila za von Willebrandov faktor (vWF) kažejo mutacijo: koagulacijski faktor je premajhen ali je okvarjen. Genska mutacija se lahko pojavi pri moških in ženskah.
Hemofilija: pregledi in diagnoza
Če ima nekdo pogosto spontano krvavitev (na primer krvavitev iz nosu) ali modrice, je to lahko znak hemofilije. Ta sum je še posebej očiten, če družina že pozna primere hemofilije. Prizadete mora zdravnik pojasniti povečano nagnjenost k krvavitvam. Prva oseba za stik, če sumite na hemofilijo, je vaš družinski zdravnik:
Zdravnik bo najprej v pogovoru z bolnikom vzel bolnikovo anamnezo (anamnezo): podrobno je opisal simptome, vprašal o morebitnih osnovnih boleznih in ali so v družini znani primeri hemofilije.
Laboratorijski testi so še posebej pomembni za razjasnitev možne hemofilije. Zdravnik bolniku odvzame vzorec krvi, da ga v laboratoriju pregleda za različne parametre: Pri hemofiliji je tako imenovani aPTT (aktiviran delni tromboplastinski čas) daljši kot pri zdravih ljudeh. Nasprotno pa sta tako imenovana hitra vrednost (tromboplastinski čas, TPZ) in plazemski trombinski čas (PTZ) na splošno normalna; podaljšajo se le pri hudi hemofiliji. Število krvnih ploščic (trombocitov) in tako imenovani čas krvavitve sta normalna tudi pri hemofiliji A in B. Pri von Willebrand-Jürgenskem sindromu se čas krvavitve podaljša. Čas krvavitve je čas, potreben za ustavitev krvavitve.
Za nedvoumno določitev hemofilije A ali B je treba analizirati aktivnost faktorjev koagulacije (VIII, IX). Za to je odgovoren specialist hematologije ali specializiran zdravstveni center (center za hemofilijo).
Hemofilija: test pri novorojenčkih in nerojenih otrocih
Če je družina že razvila hemofilijo, se koagulacija novorojenčkov moških običajno preveri takoj po rojstvu. Na ta način lahko v zgodnji fazi odkrijemo hemofilijo. Med nosečnostjo lahko preverite tudi hemofilijo.
Če ženska sumi, da ima genetsko nagnjenost k hemofiliji in je zato potencialna nosilka, je to mogoče razjasniti z genetskim testom.
Hemofilija: potek bolezni in prognoza
Hemofilija še ni ozdravljena. Bolniki se morajo skozi vse življenje spopadati s pomanjkanjem faktorjev strjevanja krvi. S pomočjo koncentratov faktorjev pa lahko običajno vodijo večinoma normalno življenje.
Če se ne zdravi, zmerna in huda hemofilija pogosto povzroči resne zaplete. Na primer, krvavitev v mišice lahko povzroči poškodbe mišic. Krvavitev v sklepih lahko povzroči osteoartritis in otrdelost sklepov. Takšnim zapletom se lahko izognemo, če hemofilijo odkrijemo zgodaj in jo dosledno zdravimo.
Tags.: spi prva pomoč paliativna medicina