Prader-Willijev sindrom
Clemens Gödel je samostojni delavec medicinske ekipe
Več o strokovnjakihja Vse vsebine preverjajo medicinski novinarji.Prader-Willijev sindrom (PWS) je posledica prirojene okvare genskega materiala. Prizadeti dojenčki so kratki, duševno nerazviti in mišičasti. V zgodnjem otroštvu razvijejo nenasitno lakoto, ki vodi do izrazite debelosti. Bolezni, ki nastanejo, je treba zdraviti s terapijo različnih medicinskih specialitet. Več o simptomih, diagnozi in zdravljenju Prader-Willijevega sindroma preberite tukaj!
Kode ICD za to bolezen: Kode ICD so mednarodno priznane kode za medicinske diagnoze. Najdemo jih na primer v zdravniških pismih ali potrdilih o nezmožnosti za delo. Q87
Prader-Willijev sindrom: opis
Prader-Willijev sindrom (napačno tudi Willi-Praderjev sindrom) so leta 1956 prvič opisali pediatri Andrea Prader, Alexis Labhart in Heinrich Willi. Približno eden od 20.000 novorojenčkov trpi zaradi Prader-Willijevega sindroma. Vzrok je genetsko sprožena disfunkcija hipotalamusa, pomembnega preklopnega centra v možganih. Manifestacija Prader-Willijevega sindroma je lahko zelo različna in zapletena.
Prader-Willijev sindrom: simptomi
Prizadete plodove opazimo že pred rojstvom. V maternici se premikajo opazno malo. Srčni utrip je nižji od običajnega. V času rojstva so plodovi s Prader-Willijevim sindromom v nenormalnem položaju v materinem telesu. Dojenčki med porodom in po njem potrebujejo veliko podpore.
Takoj po rojstvu prizadeti novorojenčki izstopajo zaradi sedečega načina življenja, (mišične) šibkosti in nizke porodne teže. Tipično kričanje po porodu je lahko tudi odsotno ali le šibko. Zaradi izrazite šibkosti in posledičnih motenj sesanja in požiranja dojenčki težko pijejo.
Dojenčki s Prader-Willijevim sindromom pogosto kažejo določene zunanje značilnosti. Za ozek obraz so značilne mandljeve oči in majhna usta s tanko zgornjo ustnico. Lobanja je pogosto dolga (dolidohocefalija), roke in noge pa majhne. Hrbtenico lahko upognemo v obliki črke S (skolioza). Kostna snov v celotnem telesu kaže poškodbe in napake (osteoporoza / osteopenija) na rentgenski sliki. Pigmentacija kože, las in mrežnice se lahko zmanjša. Možne so tudi motnje vida in škiljenje (strabizem) oči. Mošnja je majhna in pogosto prazna (nespuščeni testisi). Na splošno je razvoj bolnih otrok zakasnjen.
Ob koncu prvega leta življenja se mišična oslabelost nekoliko izboljša. Vedno pa obstaja vsaj blaga šibkost. Normalne dejavnosti lahko hitro postanejo utrudne in naporne za bolnike s Prader-Willijevim sindromom. Kljub temu otroci prav tako uživajo v tipičnih otroških dejavnostih. Rast se pri malčku znatno zmanjša.
Neomejen vnos hrane
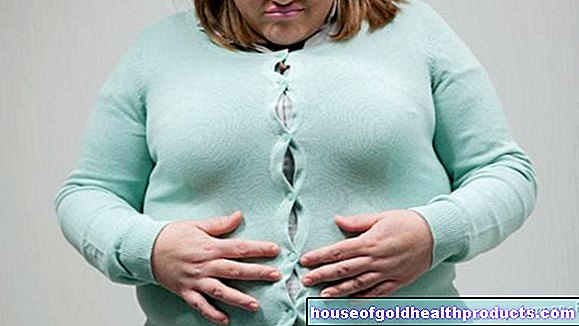
Z napredovanjem bolezni prizadeti otroci jedo vse več (hiperfagija) - brez občutka sitosti. Hrano se kopiči in tudi v hujših primerih ukrade. Otroci zelo težko nadzorujejo svoje prehranjevalne navade. Zato razvijejo izrazito debelost (debelost). Še posebej hitro pridobivajo na teži, ker obstaja izrazita razlika med visokim vnosom kalorij in nizko porabo energije.
Prekomerna telesna teža prinaša s seboj značilne sekundarne bolezni: srce in pljuča trpijo zaradi stresa zaradi debelosti. Četrtina vseh prizadetih je do 20. leta starosti razvila diabetes mellitus. Motnje spanja, venske bolezni (tromboflebitis) in zadrževanje vode so tudi med množico možnih posledic. Med potekom bolezni lahko pride do motenj spanja. Poleg ponavljajočih se prekinitev dihanja so možne motnje dnevnega-nočnega ritma ali globok spanec. Prizadeti otroci se težko učijo.
Moten je razvoj pubertete
Tipičen rast v puberteti je majhen. Prizadeti otroci običajno ne zrastejo višje od 140 do 160 centimetrov. Čeprav se puberteta lahko začne prezgodaj (prezgodnji adrenarhe), se v mnogih primerih razvoj pubertete nikoli ne konča. Prizadeti običajno ostanejo sterilni. Pri fantih penis in še posebej moda ostanejo majhni. Na skrotumu ni pigmentacije in gub. Glasovni premor morda ne bo prišel. Pri deklicah ostajajo sramne ustnice in klitoris nerazvite. Prva menstrualna krvavitev se sploh ne pojavi, prezgodaj ali pozno, včasih le med 30. in 40. letom starosti.
Duševni in duševni razvoj
Pri Prader-Willijevem sindromu je moten tako duševni kot psihomotorični razvoj. Mejniki v razvoju otrok so pogosto doseženi pozneje kot pri zdravih vrstnikih. Jezikovni in motorični razvoj včasih traja dvakrat dlje kot pri zdravih vrstnikih.
Povprečni količnik inteligence (IQ) je med 60 in 70 in je zato precej pod normo. Fizična šibkost otežuje govor. Razvoj govora zamuja, razumevanje govora pa je oslabljeno, odvisno od nizkega IQ. Približno 40 odstotkov bolnikov je na robu duševne motnje. Ne glede na IQ se pojavijo tudi učne motnje, kot so aritmetične težave. Uspešnost šole je večinoma pod povprečjem.
Opazen je lahko tako razvoj čustev kot vedenje. Prizadete ljudi včasih opisujejo kot trmaste in nagajive. Psihiatrične nepravilnosti se lahko pojavijo že v zgodnjem otroštvu: opisane so oblike tako imenovanega opozicijskega vedenja ter togo in posesivno vedenje. Rutinski procesi so lahko težki. Včasih je treba procese kompulzivno ponoviti. Avtistične lastnosti se pojavijo pri približno 25 odstotkih bolnikov. Pogosta je tudi motnja pomanjkanja pozornosti (ADD).
Simptomi se običajno povečujejo s starostjo in prekomerno telesno težo. Pri starejših odraslih pa lahko simptomi Prader-Willijevega sindroma zlahka izginejo. Približno deset odstotkov jih trpi za psihozo. Poleg tega so epilepsije in oblike "zasvojenosti s spanjem" (narkolepsija) povezane s Prader-Willijevim sindromom.
Prader-Willijev sindrom: vzroki in dejavniki tveganja
Vzrok Prader-Willijevega sindroma naj bi bil disfunkcija dela srednjih možganov, znanega kot hipotalamus. To med drugim povzroči pomanjkanje pomembnega rastnega hormona. Motnjo v približno treh četrtinah primerov povzroči pomanjkanje genskega segmenta na kromosomu 15 (15q11-q13). Pri Prader-Willijevem sindromu se zdi, da je okvara v očetovi kopiji kromosoma še posebej pomembna (70 do 75 odstotkov ), tako da obstaja samo ena kopija gena. Druga možnost je, da oba gena dvojnega niza kromosomov prihajata samo od matere (uniparentalna disomija, 25 do 30 odstotkov). Tako imenovana "napaka pri odtisu" je manj pogosta (en odstotek). Izraz "vtiskovanje" opisuje dejstvo, da se geni berejo glede na njihov izvor (na materinski ali očetovski strani).
V večini primerov motnja ni dedna. Običajno se razvije le med razvojem zarodnih celic ali po oploditvi. Po drugi strani pa je možno, da obstoječe genske mutacije (večinoma tako imenovane uravnotežene translokacije) povzročijo tudi Prader-Willijev sindrom. V teh primerih se poveča tveganje dedovanja.
Prader-Willijev sindrom: preiskave in diagnoza
Vse novorojenčke z vztrajno in nepojasnjeno šibkostjo je treba testirati na PWS. Tudi neonatalni zdravnik (neonatolog) ali pediater (pediater) bo po rojstvu zaradi vedenja otroka posumil na Prader-Willijev sindrom. Tako imenovani napovedni testi se ne izvajajo brez dokazov o prisotnosti tega sindroma. Vendar je povsem mogoče diagnosticirati Prader-Willijev sindrom že pred rojstvom.
Fizični in tehnični pregledi
V večini primerov že fizični pregled daje visoko stopnjo suma na Prader-Willijev sindrom (Holms Criteria 1993, 2001). Glavna značilnost Prader-Willijevega sindroma je izrazita šibkost, ki je še posebej očitna pri pitju. Videz daje tudi namige. Normalno zaznavni refleksi so le šibko izraženi.
Diagnostično je v pomoč tudi merljivo pomanjkanje rastnega hormona v krvi. Prizadenejo se tudi spolni hormoni (estrogen, testosteron, FSH, LH). To spremlja nerazvitost spolnih organov. Delovanje skorje nadledvične žleze je v mnogih primerih moteno. Tvorba spolnih hormonov (androgenov) se lahko začne tudi prezgodaj (zgodnji adrenarhe).
Sumljiv je lahko tudi pregled možganskih valov (elektroencefalogram, EEG).
Genetski pregled
Za potrditev suma na Prader-Willijev sindrom se opravijo genetski testi. V prvem koraku preučimo metilacijo ključne točke na kromosomu 15 (15q11.2-q13, "lokus SNRPN"). Encimi lahko na DNA vežejo tako imenovane metilne skupine in jo s tem spremenijo. V več kot 99 odstotkih ta pregled postavi diagnozo. Sicer pa se izvaja še ena običajna metoda za odkrivanje kromosomskih sprememb, fluorescenčna hibridizacija in situ (FISH).
Podobne bolezni so Martin Bell -ov sindrom ali Angelmannov sindrom. Motnja pri sindromu Martin Bell je na kromosomu X (sindrom krhkega X). Pri Angelmannovem sindromu in Prader -Willijevem sindromu je v večini primerov isti položaj na 15. kromosomu izbrisan - pri Angelmannovem sindromu pa le tisti na materinskem kromosomu.
Prader-Willijev sindrom: zdravljenje
Za Prader-Willijev sindrom ni zdravila. S strogo vodeno in podporno terapijo pa lahko simptome omilimo. Glavni sestavni deli zdravljenja so nadzor hrane, hormonsko nadomestno zdravljenje in zdravljenje vedenjskih težav.
prehrana
Zagotoviti je treba ustrezno prehrano in zadostno rast, zlasti če je mišična oslabelost huda. Za lažji vnos hrane lahko uporabite sonde ali posebne umetne bradavice. Poleg tega je treba sestaviti načrt hranjenja z dobro dokumentiranim vnosom kalorij in nadzorom teže.
Ko pa otroci odrastejo, razvijejo motnjo prenajedanja. Nato je treba upoštevati načrt s strogim omejevanjem kalorij. Omejitev maščobe ni vedno usmerjena, saj je maščoba pomembna za razvoj rogov. Motnja hranjenja lahko zahteva strogo upravljanje dostopa do hrane. Prehranjevalna motnja pri Prader-Willijevem sindromu ima še posebej resen vpliv na potek bolezni, saj se prizadeti malo gibljejo, zato obstaja velika razlika med vnosom kalorij in potrebami. Bistveno je, da starše intenzivno vključimo in bolnemu otroku ponudimo trdno strukturo.
Hkrati je treba zagotoviti zadostno oskrbo z vitamini in minerali.Pri Prader-Willijevem sindromu se pogosto pojavljajo motnje presnove kosti, ki jih je mogoče preprečiti z vnosom vitamina D in kalcija. Redno je treba spremljati razvoj okostja, zlasti hrbtenice.
Aktivnost in motorični razvoj bolnega otroka se redno pregleduje in po potrebi terapevtsko podpira s fizioterapijo ali podobnimi metodami.
Rastni hormon
Od drugega leta dalje lahko rastlinski hormon HGH dajemo tudi kot zdravilo. To terapijo je treba prekiniti, ko so rastne plošče v kosti zaprte (rentgenska kontrola). Hormonsko terapijo je treba vedno pozorno spremljati. Dajanje hormonov pozitivno vpliva na razvoj telesa, vendar so stranski učinki edem stopala, poslabšanje ukrivljenosti hrbtenice (skolioza) ali povečanje pritiska v lobanji (psevdotumor cerebri). Motnje dihanja se lahko pojavijo na začetku zdravljenja. Zato je pomembno, da na začetku terapije spremljate spanje. Spremljanje hormonske terapije s HGH vključuje tudi redne meritve ravni ščitnice in ravni rastnega faktorja IGF-1 v krvi.
V primeru motenj pubertete se lahko spolni hormoni dajejo v obliki depo injekcij, hormonskih obližev ali gela. To lahko izboljša vedenjske težave. Estrogeni prav tako pomagajo pri izgradnji kosti, vendar imajo tudi številne stranske učinke.
Psihološka podpora
Prizadeti otrok bi moral prejeti pomoč za podporo vedenjskemu razvoju in doseganju razvojnih mejnikov. Pri Prader-Willijevem sindromu je treba izobraževati zlasti socialne veščine. Spodbujati je treba tudi interakcijo z vrstniki in skrbniki. V šoli bo morda potreben nadzor enega na enega. Po potrebi je treba prilagoditi bivalno in delovno okolje. Psihiatrične nepravilnosti lahko zahtevajo zdravljenje z zdravili, na primer z antagonistom serotonina. Cilj intenzivne podpore je čim boljša neodvisnost.
Kirurške potrebščine
Vsako razpoko ustnice in nepca, ki lahko obstaja že ob rojstvu, lahko kirurgi zdravijo v zgodnji fazi. Nepravilne poravnave oči, zlasti škiljenje, lahko zdravimo tudi kirurško, da preprečimo motnje vida. Najprej lahko pomaga začasno pokrivanje zdravega očesa.
Nerazvitost spolnih organov lahko zahteva operacijo, s katero se moda premakne iz spodnjega dela trebuha v mošnjo.
Skeletne spremembe pri Prader-Willijevem sindromu lahko zdravimo tudi kirurško. S-položaj hrbtenice (skolioza) v hudih primerih zahteva kirurško korekcijo, vendar se običajno zdravi brez operacije (na primer s steznikom.
Prader-Willijev sindrom: potek bolezni in prognoza
Najzgodnejša diagnoza "Prader-Willijevega sindroma" lahko pozitivno vpliva na dolgoročno prognozo. Pozitiven vpliv na (prehranjevalno) vedenje in morebitno dajanje rastnih hormonov lahko izboljšata kakovost življenja prizadetega otroka.
Po diagnozi je treba redno preverjati prehranjevalno vedenje, težo in rast. Preverjajo se razvoj, delovanje, vedenje in psihiatrične nepravilnosti. Strogo opazovanje in oskrba specialista, ki lahko usklajuje in zagotavlja interdisciplinarno oskrbo, je zelo pomembna.
Bolniki imajo povečano tveganje za ponavljajoče se okužbe. Med operacijami je treba biti še posebej previden: Po anesteziji faza prebujanja pogosto traja dlje in obstaja povečano tveganje za motnje dihanja.
Največji problem pa je povečanje debelosti. V življenju prevladujejo nastali zapleti. Zaradi zapletov se poveča tudi smrtnost. Povečana smrtnost pri Prader-Willijevem sindromu je torej predvsem posledica bolezni srca, ožilja ali pljuč.
Tveganje dedovanja je majhno. V večini primerov je genetska sprememba spontana in se ne podeduje. Tveganje za starše drugega otroka s Prader-Willijevim sindromom je majhno. Prizadeti pogosto ostanejo brez otrok.
Le v redkih primerih je Prader-Willijev sindrom posledica sprememb genetskih informacij v naboru starševskih kromosomov, ki so dedne in vodijo tudi do večje verjetnosti dedovanja. Če ima otrok Prader-Willijev sindrom, priporočamo, da starši, ki želijo imeti otroke, poiščejo nasvet pri človeških genetikih.
Tags.: potovalna medicina gpp dojenček malček