Spinalna mišična atrofija
in Florian Tiefenböck, zdravnik Posodobljeno dneMaximilian Reindl je študiral kemijo in biokemijo na LMU v Münchnu, od decembra 2020 pa je član uredniške ekipe Seznanil se bo z medicinskimi, znanstvenimi in zdravstvenimi temami, da bodo te razumljive in razumljive.
Več objav avtorja Maximilian ReindlFlorian Tiefenböck je študiral medicino ljudi na LMU v Münchnu. Marca 2014 se je kot študent pridružilju in od takrat uredniško skupino podpira z medicinskimi članki. Po prejemu zdravniške licence in praktičnega dela na področju interne medicine v Univerzitetni bolnišnici Augsburg je od decembra 2019 stalni član ekipe in med drugim zagotavlja medicinsko kakovost orodij
Več objav avtorja Florian Tiefenböck Vse vsebine preverjajo medicinski novinarji.
Spinalna mišična atrofija ali na kratko SMA je redka bolezen, pri kateri nekatere živčne celice v hrbtenjači odmrejo. Dražljaji in impulzi iz možganov potem ne dosežejo več svojega cilja: mišic. To povzroča izgubo mišic in paralizo. Obstajajo različne oblike SMA. Najhujše se začne v otroštvu. Nova zdravljenja obljubljajo trajno izboljšanje zdravja. Več o spinalnih mišičnih atrofijah preberite tukaj.
Kode ICD za to bolezen: Kode ICD so mednarodno priznane kode za medicinske diagnoze. Najdemo jih na primer v zdravniških pismih ali potrdilih o nezmožnosti za delo. G12

Kratek pregled
- Kaj je spinalna mišična atrofija? Skupina bolezni mišične oslabelosti. Temeljijo na smrti nekaterih živčnih celic v hrbtenjači, ki nadzorujejo mišice (motorične nevrone). Zato so SMA med boleznimi motoričnih nevronov.
- Kakšne oblike obstajajo? Dedne spinalne mišične atrofije so večinoma SMA z določeno genetsko okvaro na kromosomu 5 (SMA, povezana s 5q). Zdravniki razlikujejo med štirimi različnimi oblikami: SMA tip 1 - tip 4. Poleg tega obstajajo sporadične oblike, katerih dednost ni gotova.
- Pogostost: redka bolezen; podedovana SMA prizadene približno enega novorojenčka na 7000.
- Simptomi: trzanje mišic, progresivna mišična oslabelost, izguba mišic, paraliza. Prelivi se razlikujejo glede na obliko SMA.
- Vzroki: Dedna spinalna mišična atrofija tipa 1-4 je posledica genetske okvare na kromosomu 5, natančneje na genu SMN1. Posledično telesu primanjkuje posebne beljakovine, beljakovine SMN. Ta primanjkljaj poškoduje motorične nevrone v hrbtenjači.
- Diagnoza: Genetski pregled za spremenjeno genetsko sestavo SMN, fizični pregledi, elektronevrografija, elektromiografija, krvni testi (npr. CK)
- Zdravljenje: Možna je genska nadomestna terapija ali dajanje zdravil za spajanje modulatorjev. Spremlja fizioterapijo, govorno terapijo, terapijo bolečine in psihoterapijo. Po potrebi kirurški poseg na hrbtenici. Načrt zdravljenja je odvisen od vrste SMA.
- Napoved: V primeru dednega proksimalnega SMA imajo nove možnosti zdravljenja vzročni učinek in lahko pozitivno vplivajo na potek bolezni. Začetek zdravljenja je ključnega pomena. Zdravljenje še ni na voljo vsakemu bolniku. Če se ne zdravijo, otroci s SMA tipa 1 običajno umrejo v prvih dveh letih. Pričakovana življenjska doba pri tipih 3 in 4 se skoraj ali ne skrajša.
Kaj je spinalna mišična atrofija?
Pri spinalni mišični atrofiji (SMA) nekatere živčne celice v hrbtenjači umrejo. Običajno nadzorujejo mišice, zato strokovnjaki te živčne celice imenujejo motorični nevroni. V skladu s tem SMA spadajo med tako imenovane bolezni motoričnih nevronov.
Pri spinalnih mišičnih atrofijah so prizadeti spodnji (drugi) motorični nevroni, ki so s svojimi dodatki neposredno povezani z mišicami. Zaradi poškodbe do mišic pride manj ali nič več živčnih signalov. Mišice postajajo vse šibkejše in manjše (izguba mišic / atrofija mišic).
Zdravniki razlikujejo med različnimi oblikami spinalne mišične atrofije. Daleč največja skupina je dedna SMA, pri kateri so prizadete mišice blizu trupa (proksimalno). Temeljijo na določeni genetski okvari. Približno eden od 7.000 novorojenčkov ga bo razvil.
Spinalna mišična atrofija je na splošno redka bolezen. Kljub temu je druga najpogostejša avtosomno recesivna dedna bolezen, pa tudi najpogostejši vzrok smrti dojenčkov ali malčkov zaradi genetske okvare.
Kakšne vrste spinalne mišične atrofije obstajajo?
Zdravniki razlikujejo dedne (dedne) oblike SMA od sporadičnih. Druga klasifikacija spinalne mišične atrofije se nanaša predvsem na mišične skupine, ki so bile najprej prizadete. Obstajajo
- Proksimalni SMA: S približno 90 odstotki tvorijo največjo skupino SMA. Simptomi se začnejo v mišicah blizu trupa, torej proksimalno.
- Neproksimalna SMA: Tu so najprej prizadete bolj oddaljene mišične skupine, kot so roke in stopala (distalna SMA). V nadaljevanju se lahko ti SMA razširijo tudi na mišice blizu sredine telesa.
- Posebne oblike (npr. Spinobulbarna mišična atrofija tipa Kennedy)
Proksimalna spinalna mišična atrofija
Dedne proksimalne spinalne mišične atrofije so večinoma bolezni, ki temeljijo na specifični genetski okvari (SMA, povezana s 5q, okvara na kromosomu 5). Ti pa so razdeljeni v štiri različne oblike. Razvrstitev temelji na času, ko se pojavijo prvi simptomi, in na poteku bolezni.
Spinalna mišična atrofija tipa 1: To je najpogostejša in najresnejša oblika SMA. Imenuje se tudi "Werdnig-Hoffmannova bolezen" ali "akutna infantilna SMA". Bolezen se običajno začne v zgodnjem otroštvu. Mišična šibkost vpliva na celotno telo - zdravniki govorijo tudi o "sindromu disketnega dojenčka" (iz angleščine floppy = mlahav, dojenček = dojenček, otrok). Večina nezdravljenih otrok s SMA tipa 1 umre pred starostjo dveh let.
Spinalna mišična atrofija tipa 2: Ta oblika SMA je znana tudi kot "vmesna spinalna mišična atrofija" ali "kronična infantilna SMA". Prvi simptomi se običajno pojavijo pred starostjo 18 mesecev. Prizadeti imajo včasih bistveno skrajšano pričakovano življenjsko dobo.
Spinalna mišična atrofija tipa 3: Znana je tudi kot "juvenilna spinalna mišična atrofija" ali "Kugelberg-Welanderjeva bolezen". Ta SMA se običajno začne po starosti 18 mesecev in pred zgodnjo odraslostjo. Mišična šibkost je blažja kot pri tipu 1 ali 2. Prizadeti imajo le nekoliko skrajšano pričakovano življenjsko dobo.
Spinalna mišična atrofija tipa 4: Podobna je SMA tipa 3, vendar se pojavi le v odrasli dobi (običajno> 30 let). Mišična šibkost je manj izrazita in napreduje počasneje kot pri SMA tipa 3.
Prehodi med različnimi različicami so tekoči. V nekaterih primerih to otežuje jasno razmejitev. Tudi nekatere genetske predispozicije igrajo pomembno vlogo pri resnosti zadevne bolezni.
Druge spinalne mišične atrofije
Poleg teh proksimalnih oblik obstajajo še druge oblike spinalne mišične atrofije. Sem spadajo na primer redkejše, tudi dedne distalne spinalne mišične atrofije. Pri njih se simptomi običajno začnejo v mišičnih skupinah, ki so bolj oddaljene od telesa.
V primeru občasnega SMA dedovanje ni zagotovljeno. Poleg tega ni mogoče določiti družinskega kopičenja. V literaturi to vključuje:
- Hirayama tip (juvenilna distalna SMA, bolezen okoli 15. leta, prizadene mišice rok, običajno zastane tudi brez terapije in se lahko celo izboljša)
- Vulpian-Bernhard tip (tudi sindrom "mlatilnih rok" z nastopom v ramenskem obroču, običajno od 40. leta starosti)
- Tip Duchenne-Aran (najprej prizadete mišice rok, ki se širijo proti trupu telesa, običajno po 30. letu starosti)
- Peronealni tip (sindrom "mlatilnih nog", najprej na spodnjih nogah)
- Progresivna bulbarna paraliza (motnje govora in požiranja, prizadene približno 20 odstotkov bolnikov z amiotrofično lateralno sklerozo)
Nekatere občasne oblike SMA (sindrom "mlačne roke / noge", progresivna bulbarna paraliza) se v specializiranih krogih štejejo med različice amiotrofične lateralne skleroze (ALS). Ta članek obravnava predvsem dedne proksimalne spinalne mišične atrofije.
Spinobulbarna mišična atrofija
Spinobulbarna ali bulbospinalna mišična atrofija (Kennedyjev tip, Kennedyjev sindrom) je dedna bolezen. Pogosto se začne v mladih do srednjih letih. Ta posebna oblika SMA se podeduje na X-vezan recesiven način in zato prizadene samo moške (ker imajo moški le en kromosom X, pri ženskah prevladuje drugi, zdravi kromosom X in bi nadomestil napako).
Mišična oslabelost mišic blizu telesa na nogah in rokah ali ramenih ter jezika in grla je pogosta. Posledično imajo prizadeti na primer težave z govorjenjem in požiranjem. Prav tako se pritožujejo zaradi tresenja, mišičnih krčev in trzanja. Prizadeti moški imajo pogosto tudi zakrnele testise in so sterilni. Poleg tega se mlečne žleze povečajo (ginekomastija).
Spinobulbarna mišična atrofija je običajno počasna. Pričakovana življenjska doba je skoraj omejena.
Kako prepoznate spinalno mišično atrofijo?
Za spinalno mišično atrofijo sta značilni progresivna mišična oslabelost do paralize (pareze) in trzanje mišic. Zaradi poškodbe živcev mišice ne prejemajo več električnih impulzov, zaradi česar se sčasoma krčijo (mišična atrofija). Natančni znaki in pritožbe so odvisni od ustrezne oblike. Naslednji razdelek obravnava simptome dedne proksimalne SMA.
Simptomi otroške spinalne mišične atrofije tipa 1
Pri SMA tipa 1 se simptomi pojavijo v prvih šestih mesecih življenja. Pojavi se splošna mišična oslabelost - to je tista, ki prizadene celo telo. Poleg tega se zmanjša napetost med mišicami. Zdravniki govorijo o hipotoniji.
Pri novorojenčkih se ta mišična šibkost sprva kaže v značilni drži nog, ki spominja na ležečo žabo (drža žabjih nog). Noge so upognjene, kolena nagnjena navzven, stopala pa navznoter. Tudi samostojno dviganje ali držanje glave običajno ni mogoče.
V starejši starosti otroci s SMA tipa 1 ne morejo sedeti ali hoditi sami. Mnogi otroci tudi ne morejo govoriti, saj so lahko prizadete tudi mišice jezika.
Druga značilnost spinalne mišične atrofije tipa 1 je oblika zgornjega dela telesa: mišice prsnega koša in hrbta se ne razvijajo pravilno. To daje zgornjemu delu zvonasto obliko (prsni koš). Zaradi nizkega razvoja mišic v prsih in hrbtu prizadeti zavzamejo pogrbljeno držo.
Pogosto se povečuje tudi ukrivljenost hrbtenice (skolioza). Pogrbljena naprej in pokrčena drža povzroča dodatne težave z dihanjem. Značilno je zelo hitro in plitvo dihanje (tahipneja).
Simptomi vmesne spinalne mišične atrofije tipa 2
Spinalna mišična atrofija tipa 2 običajno povzroči le simptome v starosti od sedmih do 18 mesecev. Prizadeti otroci lahko sedijo sami, običajno pa se niti ne naučijo stati in hoditi. Mišična šibkost napreduje počasneje kot pri tipu 1.
Pri SMA tipa 2 se sčasoma pojavijo simptomi, podobni tistim pri hudi otroški obliki, kot je deformacija hrbtenice. Sklepi se otrdijo zaradi skrajšanih mišic in kite (kontrakture). Drugi znaki vključujejo tresenje v rokah in trzanje mišic v jeziku.
Simptomi mladostniške spinalne mišične atrofije tipa 3
Spinalna mišična atrofija tipa 3 se običajno pojavi po starosti 18 mesecev in pred 18. Prizadeti otroci lahko samostojno sedijo, stojijo in hodijo. Mišična šibkost, zlasti v medeničnih in nožnih mišicah, povzroča hojo.
V nekaj letih se uspešnost zmanjšuje: sprva so prizadeti težko športne dejavnosti ali plezanje po stopnicah, nazadnje pa je težko nositi na primer tudi nakupovalne torbe. Spinalna mišična atrofija tipa 3 po dolgih letih otežuje ali celo onemogoča tek in vsak drug napor, tudi pri starejših.
Na splošno pa so simptomi manj izraziti kot pri dveh drugih oblikah bolezni, tipu 1 in tipu 2. Za mnoge prizadete se kakovost življenja v daljšem časovnem obdobju skoraj ne poslabša.
Simptomi spinalne mišične atrofije odraslih tipa 4
Ta zelo redka oblika progresivne izgube mišic se začne v odrasli dobi, pogosto v tretjem desetletju življenja. Na začetku so prizadete mišice nog in kolkov. Z napredovanjem bolezni se mišična šibkost razširi tudi na ramena in roke.
Klinična slika je podobna kot pri mladostniški SMA tipa 3. Vendar je progresivna mišična oslabelost še počasnejša kot pri SMA tipa 3.
Kaj povzroča spinalno mišično atrofijo?
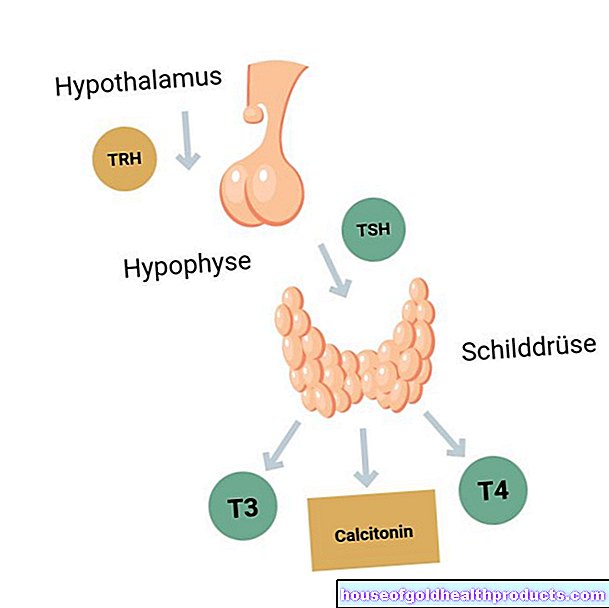
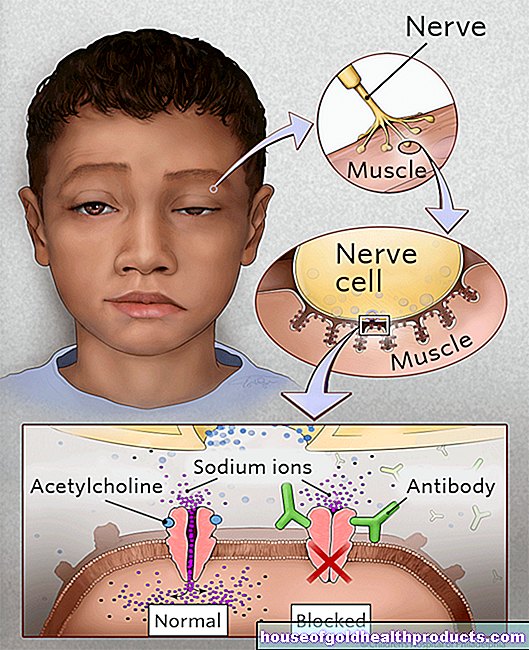
Pri spinalni mišični atrofiji umrejo drugi motorični nevroni v hrbtenjači. To so živčne celice, ki nadzorujejo mišice s svojimi dodatki. Zaradi poškodbe teh visoko specializiranih motoričnih nevronov do mišic doseže manj električnih signalov kot pri zdravih ljudeh. Če se mišične celice uporabljajo manj in so zato manj stimulirane, jih telo sčasoma razgradi.
Genetska napaka
V večini primerov je spinalna mišična atrofija dedna bolezen (dedna SMA). Vzrok za tipične proksimalne oblike SMA so napačne informacije v pacientovi genetski sestavi. Tako imenovani gen SMN1 na kromosomu 5 ni funkcionalen.
Gen SMN1 nosi informacije - torej načrt - za vitalno proteinsko molekulo, imenovano SMN. SMN pomeni "Survival (of) Motor Neuron". Brez molekule beljakovine SMN motorni nevroni sčasoma propadejo.
Res je, da je v telesu tudi soroden gen SMN2, ki načeloma lahko "kompenzira" nefunkcionalne genetske informacije SMN1. Toda to se običajno zgodi le v majhni meri. To pomeni, da izgube funkcije gena SMN1 (če je ne zdravimo) običajno ni mogoče v celoti nadomestiti z nedotaknjeno kopijo gena SMN2.
Avtosomno recesivno in avtosomno dominantno dedovanje
Genetski podatki osebe so na voljo v dveh izvodih. Posledično imajo vsi dve kopiji gena SMN1 - eno od očeta in eno od matere. Proksimalne spinalne mišične atrofije v otroštvu so običajno podedovane kot avtosomno recesivna lastnost.
To pomeni, da morata biti obe genski različici (aleli) staršev okvarjeni, da se spinalna mišična atrofija razvije pri potomcih. V primeru recesivnega dedovanja starši niso prizadeti, ker imajo poleg nefunkcionalnega tudi zdrav gen SMN1, ki kompenzira napako.
Približno vsak 45. človek je lastnik tega sistema za SMA. Par, v katerem sta oba partnerja nosilca, ima 25 -odstotno tveganje za otroka s to boleznijo.
V nekaj primerih v adolescenci spinalne mišične atrofije zlasti v odrasli dobi sledijo tudi avtosomno dominantni dediščini. V primeru prevladujoče dedovanja se pomanjkljiv gen že uveljavi - prizadeti zbolijo. Vendar to ni prej omenjena genetska napaka na kromosomu 5. Ti 5q-povezani SMA se vedno podedujejo avtosomno recesivno.
Dedovanje z drugimi oblikami SMA
Lahko se podeduje tudi neproksimalna spinalna mišična atrofija. Posebna spinobulbarna oblika (Kennedyjev tip) se deduje recesivno preko spolnega kromosoma, X kromosoma (to vpliva na genske variante, ki vsebujejo načrt za priklop mest za moške spolne hormone). V primeru občasnih oblik pa dedovanje ni zagotovljeno. Tukaj je malo znanega, zakaj ravno drugi motorični nevroni poginejo.
Pregledi in diagnoza
Diagnozo spinalne mišične atrofije običajno postavijo pediatri, pediatri, specializirani za živčne bolezni (nevropediatri) in specialisti za bolezni živčnega sistema (nevrologi). Za natančnejša pojasnila so potrebni različni pregledi. V primeru SMA so zlasti pomembni genetski testi in pregledi živcev in mišic.
Zbiranje anamneze (anamneza)
Ob vsaki bolezni zdravnik najprej vpraša o pojavu simptomov in kako je do sedaj napredoval. Pri dojenčkih in malčkih starši poročajo o spremembah in nepravilnostih v vedenju svojega otroka. Zlasti v primeru dednih bolezni se zdravniki osredotočajo tudi na anamnezo družine.
Fizični pregledi
V bistvu zdravnik s fizičnim pregledom otroka ugotovi nepravilnosti v motoričnem razvoju. Na primer preveri, ali lahko otroci samostojno držijo pokonci glave, sedijo ali samostojno premikajo roke ali noge (odvisno od starosti).
Pri starejših otrocih in odraslih s sumom na spinalno mišično atrofijo se izvajajo dodatni testi vadbe. Zdravnik preveri, koliko moči lahko zadevna oseba zbere in kako dolgo jo lahko vzdrži. Preučuje tudi vzdržljivost.
Poleg tega zdravnik testira reflekse, ki so običajno oslabljeni ali ugasnjeni, zlasti v primeru izrazitih spinalnih mišičnih atrofij. Če želite to narediti, s kladivom tapka različne kite, na primer po peti ali pod kolenom, in preveri reakcijo.
Genetske študije
Najbolj zanesljiva metoda odkrivanja (dedne) spinalne mišične atrofije je genetska analiza. Zdravniki iščejo dokaze o spremenjenem (mutiranem) genu SMN1 in številu obstoječih kopij SMN2.
Splošno pravilo je, da čim prej diagnosticiramo in zdravimo (dedno) SMA. Odvisno od oblike in razpoložljivega zdravljenja je na motorični razvoj mogoče pozitivno vplivati, preden se nepopravljivo poškodujejo motorični nevroni hrbtenjače.
Nadaljnje preiskave na SMA
Če obstaja sum na SMA, zdravniki pogosto merijo hitrost prevodnosti živcev (elektronevrografija) in mišično aktivnost (elektromiografija). Po potrebi pregledajo mišice tudi z ultrazvokom (miosonografija) ali slikanjem z magnetno resonanco (MRI).
Poleg tega zdravniki uredijo krvne preiskave. Če pride do spinalne mišične atrofije, se lahko spremenijo nekateri parametri: na primer se poveča raven kreatin kinaze (CK, tipičen mišični encim).
Zdravljenje spinalnih mišičnih atrofij
Zdravljenje spinalne mišične atrofije je zapleteno. Dolgo časa vzročna terapija ni bila mogoča za nobeno obliko SMA. Napredek medicinskih raziskav pa zdravnikom ponuja nove možnosti zdravljenja, ki v osnovi pomagajo prizadetim s proksimalno SMA (okvara gena SMN na kromosomu 5).
Poleg tega se zdravniki osredotočajo na lajšanje simptomov in zagotavljanje najboljše možne podpore prizadetim (npr. Fizioterapija, dihalna terapija, psihoterapija, po možnosti operacija).
Medicinska terapija
Novi pristopi k zdravljenju bolnikov, pri katerih SMA temelji na znani okvari gena SMN, posegajo neposredno v sam genski material ali pri nadaljnji obdelavi genskih informacij.
Cilj je pacientovemu telesu omogočiti, da samostojno proizvede zadostne količine proteina SMN, ki je ključnega pomena za motorične nevrone.
Za spinalno mišično atrofijo so na voljo naslednje možnosti zdravljenja:
- Modulatorji spajanja (Nusinersen, Risdiplam): Ta zdravila neposredno posegajo v nadaljnjo obdelavo molekul messenger RNA. Krepijo tiste procese, ki iz nedotaknjenega gena SMN2 prinašajo večjo količino beljakovin SMN.
- Gensko nadomestno zdravljenje (Onasemnogene Abeparvovec): Ta terapija posega neposredno v človeški genom. Napačno kopijo gena SMN1 v prizadetih celicah nadomesti funkcionalno genski konstrukt, dobavljen od zunaj.
Modulatorji spajanja
V primeru okvare gena SMN1 lahko telo alternativno proizvede protein SMN iz sorodnega gena SMN2. Nadomestni gen SMN2 "skoči", vendar to ni dovolj. Razlog: beljakovine v SMN2 so običajno prekratke in se hitro razgradijo.
To je posledica obdelave ustrezne SMN2 messenger RNA (SMN2 mRNA). Prenaša gradbene informacije iz genoma (DNA) na mesta proizvodnje beljakovin (ribosomi).
Če želite to narediti, najprej preberete gen SMN2 v genomu. Proizvede se predhodna sesalna RNA SMN2. Med drugim ga je treba nadalje obdelati s tako imenovanim spajanjem. Šele takrat nastane zrela messenger RNA. Posebni celični kompleksi, ribosomi, nato preberejo zrelo messenger RNA in tako proizvedejo protein SMN2. Prav ta je skrajšan in nestabilen, hitro razstavljen in ne more prevzeti funkcije SMN1.
Da bi to spremenili, aktivni sestavini Nusinersen in Risdiplam vplivata na nadaljnjo obdelavo RNA predhodnega posrednika. Posledično ti tako imenovani modulatorji spajanja na koncu povečajo količino uporabnih beljakovin SMN - in tako lahko zagotovijo ustrezno oskrbo.
Nusinersen
Zdravilo Nusinersen je tako imenovani "protismiselni oligonukleotid" (ASO). Evropska agencija za zdravila ga je odobrila leta 2017. ASO so umetno proizvedene in posebej prilagojene molekule RNA. Specifično in natančno se vežejo na Messenger RNK SMN2. To preprečuje njihovo nadaljnjo napačno obdelavo v človeški celici.
Natančneje: Nusinersen preprečuje napačno izrezovanje pomembnih informacij (ekson 7) iz RNK sporočila SMN2. Kje se nahaja ekson 7, telo nato naredi bolj funkcionalno beljakovino SMN.
Nusinersen se daje skozi ledveno punkcijo. To pomeni, da se zdravilo z brizgo injicira v hrbtenični kanal. To terapijo ponavljamo v rednih presledkih nekaj mesecev. V prvem letu zdravljenja prizadeti prejmejo šest, nato tri odmerke na leto.
Bolniki običajno dobro prenašajo zdravilo. Nusinersen vodi k ugodnejšim potekom bolezni. Študije so pokazale, da se mobilnost pri mnogih bolnikih izboljša: v mnogih primerih je bilo mogoče prosto sedeti in samostojno obračati telo. Neželeni učinki in zapleti med drugim temeljijo na ledveni punkciji (npr. Glavobol, okužbe možganskih ovojnic).
Risdiplam
Evropska komisija je marca 2021 odobrila zdravilo Risdiplam kot tretje zdravilo proti SMA, povezanemu s 5q (tipi 1-3 ali ena do štiri kopije gena SMN2). Risdiplam se jemlje vsak dan v obliki raztopljenega praška. Natančen odmerek se izračuna glede na starost in telesno težo.
Za razliko od Nusinersena Risdiplam ni "protismiselni oligonukleotid", ampak majhna molekula. Ta molekula se veže na messenger RNA za beljakovine SMN2 in jih na ta način stabilizira. Posledično nastanejo bolj funkcionalni proteini SMN.
Pogosti neželeni učinki zdravila Risdiplam so nelagodje v prebavilih, izpuščaj, zvišana telesna temperatura in okužbe sečil.
Gensko nadomestno zdravljenje
Drug pristop k zdravljenju proksimalne spinalne mišične atrofije temelji na tako imenovanem nadomestnem genskem zdravljenju. Pomanjkljiv gen SMN1 - izhodišče progresivne SMA - se "nadomesti" z novo funkcionalno kopijo gena.
Zdravilna učinkovina Onasemnogene Abeparvovec (AVXS-101), ki deluje na tem principu, je maja 2020 od Evropske agencije za zdravila (EMA) prejela pogojno dovoljenje za promet z malčki in otroki.
Po podatkih EMA se zdravilo lahko uporablja za SMA tipa 1. Pri vseh drugih oblikah bolezni SMA genetske značilnosti (število kopij SMN2) odločajo, ali je nadomestna genska terapija možna.
Z Onasemnogenom Abeparvovec se funkcionalna kopija človeškega gena SMN1 vnese v prizadete celice hrbtenjače in možganskega debla. To počnejo nekateri virusi, ki služijo kot "trajekti" za nov genski material-tako imenovani adeno-povezani virusni vektorji (vektorji AAV).
Konstrukti vektorskih genov se dajejo enkrat kot infuzija skozi veno v krvni obtok in se od tam porazdelijo po telesu. Zaradi še ne povsem razvite krvno-možganske pregrade pri majhnih otrocih lahko ti vektorji prodrejo tudi v tkivo hrbtenjače.
S prednostno vezavo teh vektorjev na posebne površinske strukture motoričnih nevronov prednostno prevzamejo genski material, da nato neodvisno proizvedejo beljakovine SMN.
Zdravljenje lahko izboljša motorične funkcije in vodi do trajnega razvojnega uspeha (npr. Sedenje, plazenje in hoja brez podpore).Med zdravljenjem se lahko vrednosti jeter včasih znatno povečajo, vendar se lahko število krvnih ploščic zmanjša. Pogosta sta tudi vročina in bruhanje. Za zmanjšanje stranskih učinkov bolnikom nekaj tednov dajemo kortikosteroide ("kortizon").
Starostno primeren motorični razvoj je na splošno mogoč le, če se je genska terapija začela presimptomatsko. Zdravljenje poteka v specializiranih centrih za živčno -mišično zdravljenje.
fizioterapija
Fizioterapija je še vedno pomemben steber zdravljenja SMA, zato vseh oblik SMA ni mogoče zdraviti z novimi pristopi zdravljenja. Redna vadbena terapija je namenjena ohranjanju telesnih sposobnosti in upočasnitvi razpada mišic.
Fizioterapevt pasivno premika dele telesa, ki so že ohromljeni. Aktivne gibalne sekvence so nato usposobljene za podporo gibljivosti in moči mišic. Pomagajo lahko tudi masaže ali toplotni in hladni tretmaji. Ti služijo tudi za sprostitev in v določenih okoliščinah upočasnijo nadaljnjo degeneracijo.
Logopedska terapija
V nekaterih primerih SMA vpliva na govorne in požiralne mišice. Potem pomaga logopedska vaja. Otroke spodbuja k učenju govora. Tudi pri starejših bolnikih lahko to običajno upočasni poslabšanje govora. Logopedi trenirajo tudi pravilno požiranje.
Tako fizioterapevti kot logopedi podpirajo prizadete z usmerjeno dihalno terapijo.
Zdravljenje za lajšanje bolečin
Bolečina ima pomembno vlogo, zlasti v naprednejših fazah bolezni. Zdravniki uporabljajo zdravila proti bolečinam, da zmanjšajo trpljenje prizadetih.
operacija
Ker lahko spinalna mišična atrofija povzroči hudo ukrivljenost hrbtenice (skoliozo), zdravniki včasih razmišljajo o operaciji. Pri tem posebej utrdijo hrbtenico.
To daje prizadetim (določeno) dodatno stabilnost trupa, ki ne omogoča le pokončnejše drže, ampak tudi ščiti kosti in sklepe. Operacija hrbtenice lahko pomaga tudi pri progresivnih težavah z dihanjem.
Psihoterapevtska oskrba
Nevromišične bolezni, kot je spinalna mišična atrofija, predstavljajo velik psihološki stres.Pacienti in svojci diagnozo obdelujejo na individualnih in skupinskih sejah, ki jih vodi psihoterapija, in razvijajo strategije za boljše obvladovanje bolezni.
Pomembno podporo ponujajo tudi skupine za samopomoč in predstavniki pacientov. Obveščajo, svetujejo in podpirajo prizadete in njihove svojce pri spopadanju z izzivi bolezni SMA.
Možnosti okrevanja po spinalnih mišičnih atrofijah
Če pride do spinalne mišične atrofije, je napoved odvisna predvsem od ustrezne oblike. Bolj ko se pojavijo simptomi, boljši je potek. Poleg tega prej zdravniki diagnosticirajo spinalno mišično atrofijo, prej lahko začnejo ustrezne ukrepe zdravljenja, še preden so bili nepopravljivo poškodovani motorični nevroni.
Nove možnosti zdravljenja z modulatorji spajanja in gensko nadomestno terapijo imajo velik potencial pri zdravljenju proksimalne SMA - zlasti na začetku (zelo) zgodaj. Podatki za zanesljivo dolgoročno prognozo pa še niso znani. Le nadaljnje študije in tesna opazovanja varnosti drog lahko zagotovijo dodatno gotovost v naslednjih (mesecih in) letih. Z novejšimi zdravili je vsaj možno dolgoročno obvladovanje bolezni ali celo ozdravitev.
SMA tipa 1 je na splošno resna bolezen. Otroci, ki razvijejo SMA tipa 1, imajo (nezdravljeno) zelo omejeno pričakovano življenjsko dobo. Hitro naraščajoča mišična oslabelost po vsem telesu vpliva tudi na dihanje. Posledice so akutna pljučnica in celo odpoved dihanja. Prizadeti otroci umrejo v prvih nekaj letih življenja.
Napoved za SMA tipa 2 je nekoliko boljša. Pričakovana življenjska doba je odvisna od resnosti bolezni: nekateri umrejo v otroštvu, večina pa jih doseže mladost. Prej ali slej - če želimo - je treba dihanje podpreti v hujših oblikah. Prizadeti ljudje ostanejo mobilni s pomočjo invalidskega vozička.
Pri SMA tipa 3 je napoved bistveno boljša - še posebej, če se prvi simptomi pojavijo pozno. Učinkovitost se v nekaj letih postopoma slabša. V starosti bo morda potreben invalidski voziček ali celo stalna oskrba. Pričakovana življenjska doba ni omejena s spinalno mišično atrofijo tipa 3.
Odrasla spinalna mišična atrofija (tip 4) je celo počasnejša od tipa 3. Ljudje imajo običajno običajno pričakovano življenjsko dobo.